When it comes to medical devices, patient safety and regulatory compliance leave no room for error. Unlike standards focused on information security (such as ISO 27001), ISO 13485 is tailored specifically to the medical device industry. It sets requirements for a quality management system (QMS) to ensure devices consistently meet both regulatory obligations and customer expectations for safety and performance.
In 2025, ISO 13485 is more than just a certification for medical device manufacturers and their suppliers — it’s often the gateway to market access. Regulatory shifts are making it indispensable:
- EU MDR 2017/745 requires manufacturers to demonstrate conformity with ISO 13485.
- FDA’s QMSR alignment means the U.S. FDA is aligning its Quality System Regulation (QSR) with ISO 13485, raising the compliance bar for companies selling into the U.S. market.
For organizations with global supply chains, this makes achieving and maintaining ISO 13485 both critical and complex. Simply put, if you’re making or supplying medical devices in 2025, you’ll need ISO 13485 to compete, to comply, and to earn trust. This blog breaks down what ISO 13485 is, why it matters, and how platforms like Scrut can help streamline parts of the journey.
What is ISO 13485?
ISO 13485 is an internationally recognized standard that defines the requirements for a quality management system (QMS) in the medical device industry. Its purpose is to ensure organizations can consistently design, develop, manufacture, install, and service medical devices that are safe, effective, and compliant with global regulations.
While it builds on the quality management principles of ISO 9001, ISO 13485 adds layers of regulatory specificity tailored for medical devices, such as risk management, traceability, and post-market surveillance.
Key focus areas of ISO 13485 include:
- Risk management throughout product design, development, and production
- Design and development controls to ensure device safety and performance at each stage
- Validation of processes and software that support medical devices
- Traceability of products and documentation across the supply chain
- Post-market surveillance and reporting to monitor performance and catch issues after launch
For example, if you’re producing a cardiac stent, ISO 13485 requires you to document the entire design history file (DHF), validation records, sterilization reports, and complaint-handling processes. Regulators will check this trail during audits.
Why is ISO 13485 important in 2025?
Medical devices leave no room for error, as even small flaws can put patient safety at risk. ISO 13485 sets the global benchmark to ensure safety and quality are built into every stage of the device lifecycle.

- Regulatory pressure is increasing: Many regions, including the EU under MDR 2017/745 and the U.S. (with FDA’s QMSR alignment), recognize or require ISO 13485 compliance.
- Global supply chains add complexity: With multiple suppliers involved in design and manufacturing, ISO 13485 ensures uniform quality across all partners and stages of production.
- Market access depends on it: For many tenders, contracts, and product registrations, ISO 13485 certification is mandatory to even enter the market.
- Trust and reputation: Certification signals to regulators, partners, and patients that quality is embedded in every stage of the device lifecycle.
- Cost of non-compliance: Failing to meet ISO 13485 can lead to regulatory penalties, rejected product approvals, recalls, delayed market entry, and reputational damage. These costs often outweigh the investment required to stay compliant.

ISO 13485 vs ISO 27001: how are they different?
It’s common for organizations to confuse ISO 13485 with ISO 27001, since both are international standards developed by ISO. However, their objectives and applications are very different:
Key takeaway: ISO 13485 is about product quality and patient safety, not information security. ISO 27001, on the other hand, is centered on establishing an ISMS to secure sensitive data. The two can overlap. For example, imagine a cloud-connected glucose monitor:
- ISO 13485 governs the device’s quality, including supplier traceability, design validation, and defect CAPA logs.
- ISO 27001 governs the data the device collects, ensuring patient health information is protected against cyber risks.
Because of this natural intersection, many medtech companies pursue both certifications together to satisfy regulators and reassure patients and partners.

What are the requirements of ISO 13485?
ISO 13485:2016 specifies structured requirements that organizations must meet in order to build, maintain, and demonstrate an effective quality management system (QMS) for medical devices. These requirements cover six main areas:
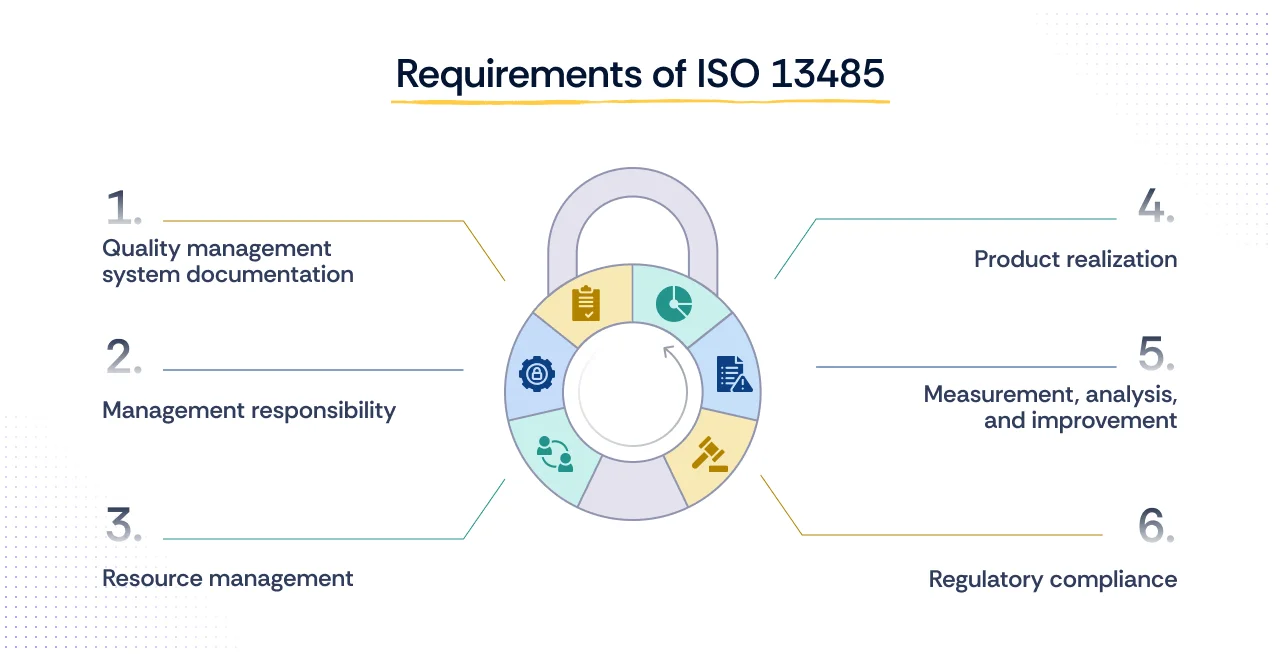
1. Quality management system documentation
Organizations must create and maintain a documented QMS, including a quality manual, policies, procedures, and records. This documentation provides consistency, traceability, and evidence for audits.
2. Management responsibility
Top management must take ownership of the QMS by defining accountability, setting measurable quality objectives, conducting reviews, and ensuring regulatory alignment.
3. Resource management
Adequate resources must be provided, qualified personnel, training, infrastructure, and a safe working environment, to ensure devices are developed and produced under controlled conditions.
4. Product realization
From design through delivery, organizations must apply risk-based controls, design validation, and verification processes to ensure products meet intended use and patient safety requirements.
5. Measurement, analysis, and improvement
The QMS must include systems for monitoring performance, conducting internal audits, managing corrective and preventive actions (CAPA), and driving continual improvement.
6. Regulatory compliance
Organizations must prove that their QMS meets applicable national and international medical device regulations, such as the EU MDR or FDA requirements, in addition to ISO 13485.
Together, these requirements ensure that quality management is systematic, auditable, and repeatable, not left to individual judgment or ad hoc practices.
Steps to achieve ISO 13485 certification
1) Scope and plan
Define what your certificate will cover: products, processes, and sites. Identify the regulatory contexts you operate in (e.g., EU MDR, US market) because they shape your QMS scope. Build a realistic plan: owners, milestones, and what “done” means for each strand (design controls, suppliers, production, complaints, CAPA, training, document control).
What you should have in hand: a scoped statement, a RACI/owners map, a remediation plan tied to clauses, and a timeline that includes internal audits and management review before certification.
2) Baseline review (gap analysis)
Compare how you currently operate against ISO 13485 requirements. Go deeper than “we have a procedure”. Test whether people actually use it, whether records exist, and whether outputs meet the intent (e.g., risk files that drive design decisions, not just risk registers for show).
Watch for gaps here: supplier evaluation and re-evaluation, validation of software used in production or QMS, calibration/maintenance control, complaint handling links to CAPA, and traceability through DHF/DMR/DHR.
3) Build or refine the QMS (Quality Management System)
Write or update the core documents so they reflect how work really happens. Keep the quality manual lean and make procedures unambiguous. Establish the record set you’ll rely on during audits i.e. forms, logs, and templates for:
- Design & development (inputs, reviews, verification/validation, change control)
- Risk management integrated into design decisions
- Supplier control (qualification, monitoring, re-qualification)
- Production and service provision (including process/sterilization/software validation where applicable)
- Identification and traceability
- Nonconformance, complaints, and CAPA
- Training & competence
- Document/record control
Output to aim for: a coherent “document tree,” and templates for DHF (design history), DMR (how to make it), and DHR (proof you made it that way).
4) Operate the system (generate real evidence)
Run the QMS. Train people, execute procedures, and let records accumulate from real work. Auditors expect to see a period of operation, not a one-week sprint. Treat change requests, supplier approvals, design reviews, calibrations, and complaint investigations as chances to produce strong, linked evidence.
A practical benchmark: many organizations target 8–12 weeks of live records before Stage 1, so sampling at Stage 2 tells a convincing story.
5) Internal audit and management review (no rubber stamps)
Audit the full scope and not just easy areas. Sample across departments and sites, and follow threads end-to-end (e.g., a complaint → investigation → CAPA → effectiveness check). Raise nonconformities honestly and drive root-cause CAPA. Management then reviews the system’s performance, resourcing, risks, changes, and quality objectives, and minutes that review with decisions and actions.
Good signal you’re ready: internal findings are closed or actively in CAPA, metrics are understood (not just reported), and managers can speak to risks and resources without coaching.
6) Stage 1 audit (readiness and completeness)
An accredited certification body reviews your documented QMS and confirms you’re ready for Stage 2. Expect questions on scope, mandatory procedures, interfaces to suppliers, and how you ensure records are controlled. You’ll get findings (if any) and a Stage 2 plan.
Outcome to chase: only minor gaps, with clear closure actions scheduled before Stage 2.
7) Stage 2 audit (does it work in practice?)
Auditors test implementation by sampling device files, production records, training, complaints, CAPA, supplier files, calibrations, maintenance, and validation evidence. Findings are categorized (typically major/minor). Majors must be corrected and verified; minors need a credible plan and follow-through.
What wins the day: consistent records that tell a linked story, staff who can explain their process, and CAPA that fixes causes—not just symptoms.
8) Certification decision, surveillance, and recertification
If you clear Stage 2 (and close any majors), your certificate is issued. It’s usually valid for three years, with annual surveillance to confirm you’re sustaining the system. At the end of the cycle, a recertification audit renews the certificate.
How to stay ready: keep audits on a rolling calendar, keep CAPA moving, review the QMS when products, suppliers, or regulations change, and keep training current.
Common challenges organizations face in ISO 13485 compliance
While the steps appear linear, most companies encounter recurring hurdles along the way:
- Heavy documentation requirements – ISO 13485 demands extensive records throughout the product lifecycle, from design files to complaint handling logs. Many organizations underestimate this effort.
- Supplier and contractor integration – Medical device companies often work with global suppliers. Aligning all of them under a single QMS is complex, especially when compliance standards differ across regions.
- Evolving regulations – Standards like ISO 13485 are harmonized with global laws such as EU MDR and FDA 21 CFR Part 820. Keeping pace with frequent regulatory changes adds ongoing pressure.
- Audit readiness – Companies that wait until just before audits to prepare face last-minute stress. The real challenge is maintaining compliance year-round as a business-as-usual practice.
- Balancing compliance with innovation – In the fast-moving medtech space, organizations often worry that strict processes may slow innovation. The key is designing a QMS flexible enough to enable both compliance and agility.
How Scrut helps with ISO 13485 compliance
With the FDA aligning U.S. regulations to ISO 13485 starting in 2026, medical device companies can’t afford gaps in their compliance programs.
Scrut helps you anticipate and prepare for these changes by streamlining documentation, automating evidence collection, and giving you continuous visibility across your compliance posture.
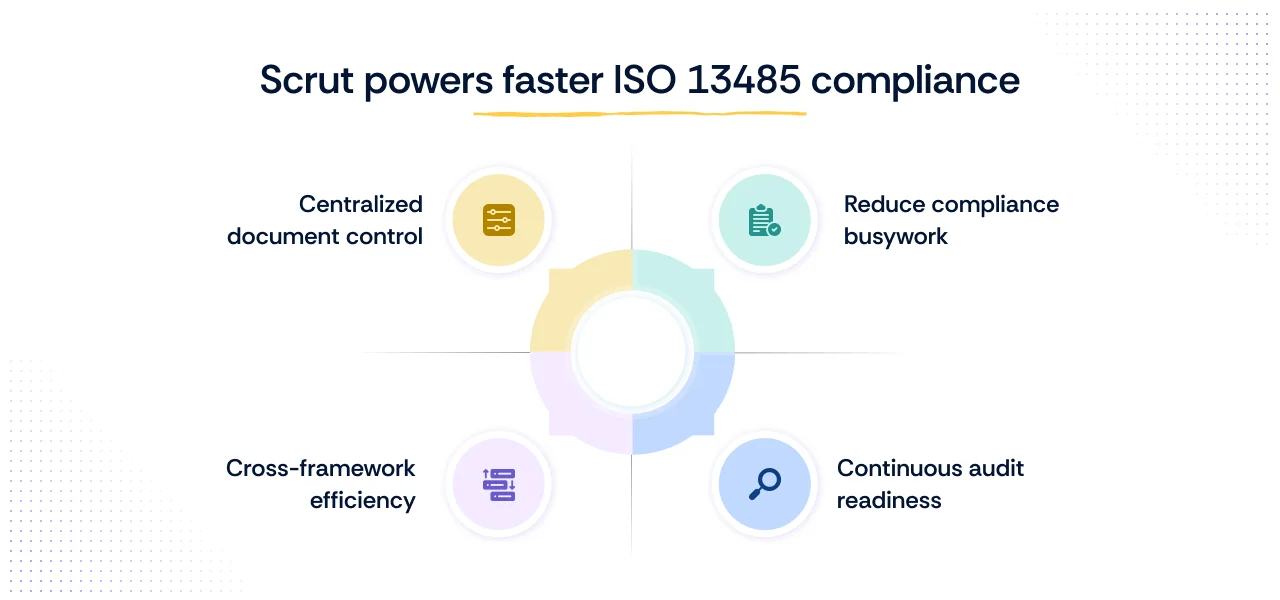
- Centralized document control: Store and manage policies, procedures, and records with version history, approvals, and templates to support QMS documentation needs.
- Cross-framework efficiency: Get pre-mapped controls across ISO 13485, ISO 27001, HIPAA, and 60+ others to avoid duplication of effort.
- Reduce compliance busywork: Automate up to 70% of manual compliance tasks like evidence collection and control monitoring via automated tests across 100+ integrations.
- Continuous audit readiness: Use a unified dashboard and continuous control testing to stay on top of compliance tasks, risks, and evidence—so audits don’t derail day-to-day work.
For medical device companies, this means less manual effort, fewer silos, and a more reliable state of readiness, especially relevant as the FDA’s QMSR will align U.S. requirements with ISO 13485 starting February 2, 2026.
Final thoughts
Achieving ISO 13485 certification is not just about passing an audit, it’s about building trust with regulators, customers, and most importantly, patients. The process, however, can feel overwhelming with documentation, evidence gathering, and ongoing compliance requirements.
This is where Scrut makes a real difference. By automating manual compliance tasks, centralizing risk and audit workflows, and providing continuous visibility across frameworks, Scrut helps medical device companies simplify ISO 13485 compliance while staying focused on innovation and growth.
If your organization is preparing for ISO 13485 certification or looking to strengthen its compliance posture, now is the right time to see how automation can transform your journey.

ISO 13485 is an international standard that defines the requirements for a quality management system (QMS) for medical devices. It applies to medical device manufacturers, suppliers, and service providers involved in designing, producing, installing, or servicing medical devices.
ISO 13485 is a voluntary standard, but it is often required to access major markets. Regulations such as the EU MDR 2017/745 and the U.S. FDA’s QMSR alignment rely on ISO 13485 principles, making certification essential for many medical device companies.
ISO 13485 requires organizations to implement a QMS covering documentation, management responsibility, resource management, product realization, performance monitoring and improvement, and regulatory compliance.
ISO 13485 focuses on quality management and safety of medical devices, while ISO 27001 focuses on information security and data protection. Many medtech companies adopt both standards because devices often handle sensitive patient data.
Certification timelines vary by organization. The process typically involves gap analysis, QMS implementation, internal audits, and two certification audits (Stage 1 and Stage 2) conducted by an accredited body.







.png)